
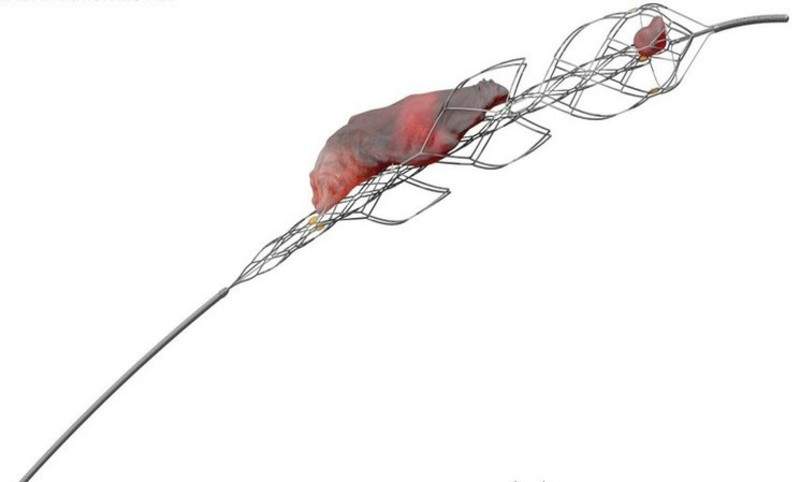
Johnson & Johnson (J&J) subsidiary Cerenovus has received approval from the US Food and Drug Administration (FDA) for its EMBOTRAP II Revascularization Device to treat ischemic stroke.
The device has been designed as a stent retriever that can be used to capture and remove life-threatening blood clots within the neurovasculature of the brain after a stroke.
Go deeper with GlobalData

Discover B2B Marketing That Performs
Combine business intelligence and editorial excellence to reach engaged professionals across 36 leading media platforms.
It restores blood flow quickly and requires minimal compression, in turn avoiding further complications.
The mechanical thrombectomy device is approved for use within eight hours of onset of stroke symptoms in patients not eligible for intravenous tissue plasminogen activator (IV t-PA) or failed IV t-PA therapy.
Cerenovus worldwide president Daniella Cramp said: “EMBOTRAP II is the product of deep collaboration between engineers and clinicians to better understand the science of blood clot, what causes them to form and how a mechanical thrombectomy device can interact with them to help improve outcomes.
“Cerenovus is committed to advancing treatment with evidence-based solutions so that fewer and fewer people are affected by the ravages of stroke.”

US Tariffs are shifting - will you react or anticipate?
Don’t let policy changes catch you off guard. Stay proactive with real-time data and expert analysis.
By GlobalDataThe FDA decision comes after a review of the ARISE II study performed to evaluate the EMBOTRAP II device in 228 patients with large vessel occlusions and moderate to severe neurological deficits.
Data revealed restoration of blood flow in 80% of subjects treated within three passes and in around 50% of those within a single pass. More than two-thirds of the participants were found to be functionally independent at 90-day follow-up.
The device also secured the European regulatory approval, where it has been used to treat more than 3,000 patients.

