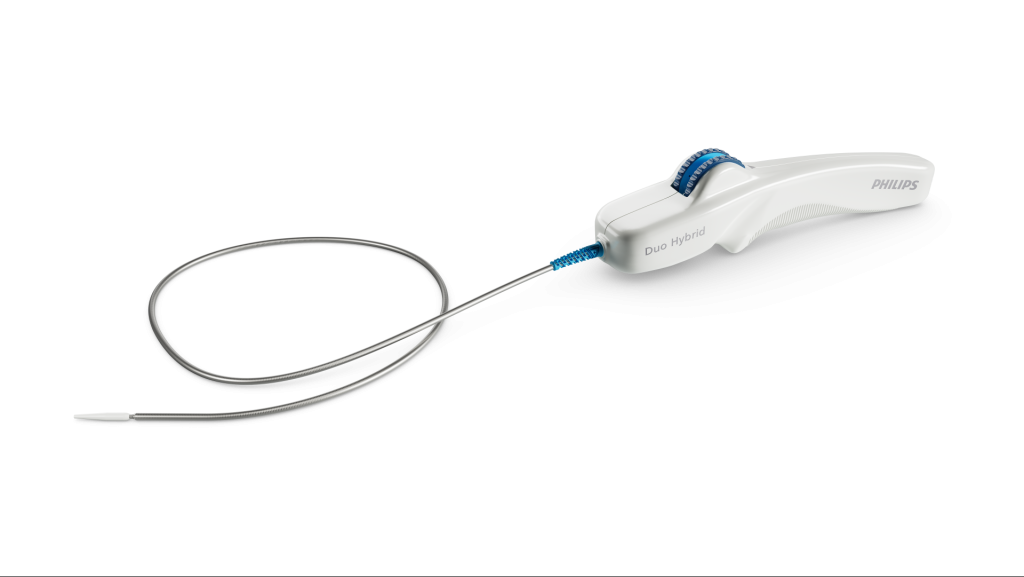
Teleflex and its subsidiary Arrow International have initiated a recall of specific intra-aortic balloon (IAB) catheter kits due to a manufacturing defect that may prevent full balloon inflation, posing a risk of serious injury or death.
The US Food and Drug Administration (FDA) has classified the recall of Arrow FiberOptix and UltraFlex IAB catheter kits as a Class I recall.
The products being recalled have codes ranging from IAB-05830-LWS to IAB-06850-U.
A total of 16,959 devices have been recalled in the US, distributed from 07 May 2022 and 8 April 2024.
The IAB catheter kits are typically used in conjunction with a balloon pump for patients undergoing cardiac and non-cardiac surgery, as well as for treating adults with acute coronary syndrome or heart failure complications.
The recall was prompted by the discovery of a manufacturing error that could cause the catheter's balloon to become overtwisted, potentially leading to incomplete inflation, blood backup, helium leakage, and difficulties during insertion.
Teleflex and Arrow International have received 322 complaints regarding this issue, including reports of 31 injuries and three deaths potentially related to the defect.
In response, the company issued an Urgent Medical Device Notification on 29 April 2024 to its customers, advising healthcare professionals to have a backup intra-aortic balloon catheter available and to inspect all catheters for signs of overtwisting balloon wrap or bent balloon shaft before usage.
The notification also recommended the use of fluoroscopic guidance during insertion and for assessing balloon inflation.
It highlighted the importance of responding to pump alarms and bedside indicators that may signal the catheter is not performing as expected.
Earlier this month, the FDA issued a Class I recall for a piece of Medtronic software used in locating anatomical structures during brain surgeries after it was found to display faulty text that potentially misleads surgeons.