

The European Commission has made changes to rules governing the provision of electronic instructions for use (eIFUs) for medical devices (MDs), as originally laid down in Regulation 207/2012.
The original legislation allowed manufacturers to provide paper-based instructions for use (IFUs) for MDs and associated software electronically, usually on digital hardware (USB sticks) or through a website.
The latest update, Implementing Regulation 2021/2226, repealed and replaced Regulation (EU) 207/2012, which was published in 2012 to allow the provision of eIFUs.
IFUcare is a pioneer in e-labeling and eIFU services that meet and exceed EU, FDA and other international legislation since 2008. As a regulatory expert, IFUcare always stays ahead of proposed regulatory updates. As such, they immediately applied the new EU provisions to their services and informed every customer of this change.
IFUcare began evaluating the forthcoming impact of the new regulation while it was still being drafted. Thanks to this head start and the flexibility of IFUcare’s platform and services, they ensured that their customers continued to benefit from having the latest legislation applied to their medical device information portals seamlessly.
Paper IFUs and eIFUs
IFUs and eIFUs are essential in ensuring medical devices are used safely, as they give users access to critical information about MDs. Therefore, the governing rules require that the level of safety, longevity and security for eIFUs is greater than, or at least equal to, those for IFUs.
IFUcare monitors all current and proposed legislation, both in the EU and globally, and its eIFU platform and associated services have been designed to make and apply changes quickly.
The eIFUs for medical devices are now widely provided, with obvious benefits: a huge reduction of paper waste and costs. Moreover, it overcomes the logistical challenges of paper IFUs such as stock management and keeping up to speed with IFU updates. But setting up a compliant eIFU solution is a complicated process as well. IFUcare makes this process simple.
Impact on manufacturer
Manufacturers that want to continue using eIFUs under the Medical Devices Regulations (MDR) framework should review the new Implementing Regulation 2021/2226 and examine how their current procedures align with the new requirements.
Although no major changes were made to the list of devices that can make use of eIFUs, the new regulations that allow manufacturers to provide eIFUs are:
- Implantable and active-implantable medical devices and their accessories
- Fixed installed medical devices and their accessories
- Medical devices and their accessories that are fitted with a built-in system to display the instructions for use
Manufacturers may provide instructions for use in electronic form instead of in paper form for the listed devices only under the following conditions:
- The devices and accessories are intended for exclusive use by professional users
- The use by other persons is not reasonably foreseeable
For software, manufacturers may provide eIFUs within the software itself.
New rules
First of all, it is important to know why the eIFU regulation needed an update. The ‘old’ eIFU rules were defined in terms of medical devices placed on the market in times of the (Active Implantable) Medical Device Directives ((AI)MDD) [1]. Since 2017, the (AI)MDD was replaced by the Medical Device Regulation (MDR) [2]. Since the MDR introduced much more and stricter rules, the associated regulations, such as the eIFU regulation, were in urgent need to be revised as well.
One of the rules introduced in the MDR is that as well as the documentation, which is shared with the healthcare professional, all implant manufacturers must also provide implant cards and information to the patient. According to the regulation, that patient information must be shared ‘by any means that allow rapid access to that information […]’. It is clear that eIFU would be ideal to serve this purpose.
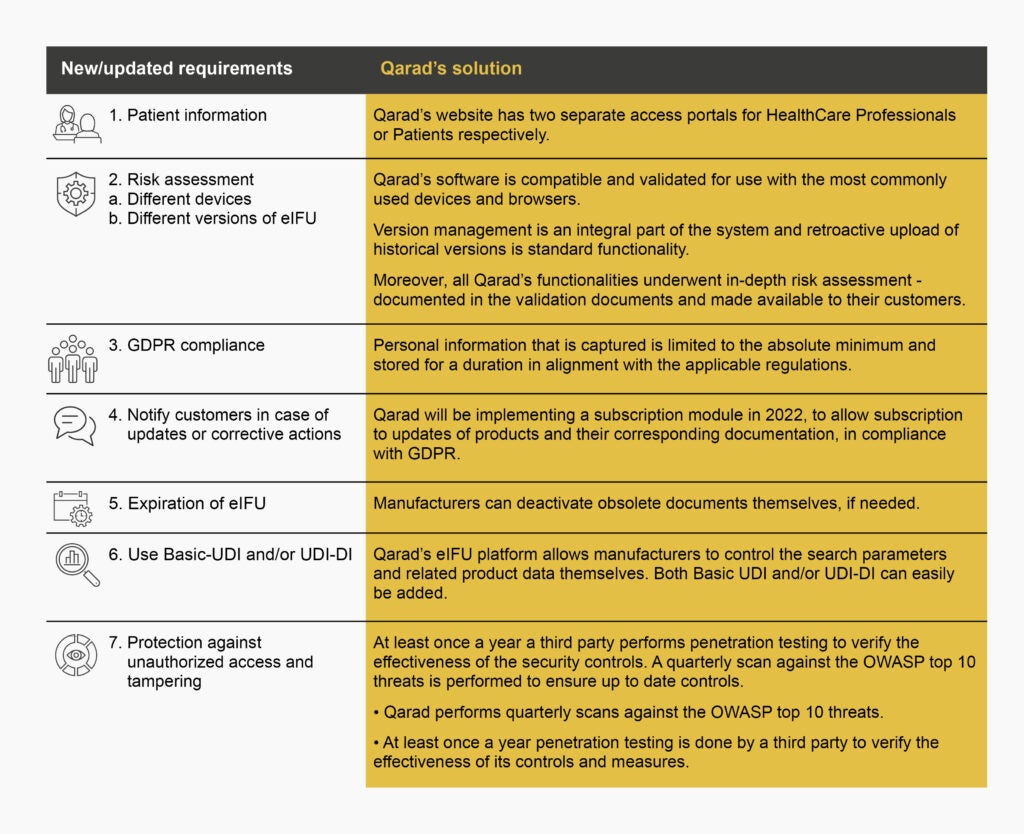
IFUcare’s eIFU website allows manufacturers to distribute both information for healthcare professionals as well as patients. Both are hosted on the same website, which facilitates the data management for the manufacturer while maintaining a clear distinction between respective HCP and Patient portals so customers can easily obtain the documents relevant to them.
Transparency is another requirement under the new regulation. Effective systems and procedures are required to be in place to ensure that device users, having downloaded an eIFU from the website, can be informed of updates or corrective actions with regard to their particular device.
IFUcare will be implementing a subscription module during 2022, which will allow end-users to subscribe to eIFU updates and corresponding documentation easily. This new feature will be aligned with the EU’s General Data Protection Regulation (GDPR) regulations.
IFUcare’s eIFU website will be explicit in consent being obtained for processing personal data. Personal information that is captured will be limited to the absolute minimum and stored for a duration in alignment with the applicable regulations.
Requirements of Article 5 of Regulation 2021/2226 state that for devices with a defined expiry date, except implantable devices, the manufacturer must keep the eIFU available for users for 10 years after the last device has been placed on the market, and at least two years after the end of the expiry date of the last produced device.
For devices without a defined expiry date and implantable devices, the manufacturer must keep the IFUs available for the users in electronic form for 15 years after the last device has been placed on the market.
All issued historical versions of eIFUs must be available on websites. As such, version information is essential. On IFUcare’s website, version management is an integral part of the system and retroactive upload of historical versions is standard functionality.
Article 6 (3) addresses the elements to be contained in the information on how to access eIFUs. Where before, regulation 207/2012 required a unique reference and any other information needed by the user, to identify and gain direct access to the appropriate eIFU, the new Regulation requires the Basic UDI-DI and/or the UDI-DI (Unique Device Identification – Device Identifier)of the device, and any additional information allowing the identification of the device, including its name and if applicable the model.
IFUcare’s eIFU platform customers have access to built-in configurable data fields they can add to their portals to post this kind of additional critical information for their end-users.
Article 7 (2) adds two new requirements for websites through which eIFUs will be provided:
- protected against unauthorised access and tampering of content in accordance with Article 4(1), point (e)
- Regulation (EU) 2016/679 (GDPR)
Security is a critical component of the infrastructure and design of the IFUcare eIFU System. IFUcare performs quarterly scans against the OWASP (Open Web Application Security Project) top 10 threats and, at least, annual penetration testing by a third party to verify the effectiveness of its controls and measures.
IFUcare’s eIFU platform and service have been in operation for more than 13 years. The technology and capabilities of their system evolve continuously, thus ensuring their clients benefit from operating on the latest hardware and software platforms.
Further information
For more details, please visit https://ifucare.io/
[1} Respectively COUNCIL DIRECTIVE of 20 June 1990 on the approximation of the laws of the Member States relating to active implantable medical devices (90/385/EEC) and COUNCIL DIRECTIVE 93/42/EEC of 14 June 1993 concerning medical devices
[2] Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, amending Directive 2001/83/EC, Regulation (EC) No 178/2002 and Regulation (EC) No 1223/2009 and repealing Council Directives 90/385/EEC and 93/42/EEC
